Some informal notes on the methods implemented in wgd¶
| Author: | Arthur Zwaenepoel |
|---|
Contents
Introduction¶
wgd is a python package and command line suite for analyzing ancient whole
genome duplications (WGDs) in genomic data sets. The main focus of wgd is
on the construction of whole paranome (the collection of all duplicate genes in
a genome) KS distributions, but it also provides tools for the inference of
intragenomic synteny and anchor points as well as downstream analyses and
visualizations of whole paranome KS distributions. On this page we provide
some background information and informal explanations about the main functionality
and methods, namely the inference of these so-called KS distributions. This
is not a tutorial on how to use wgd, for that, please have a look at the
command line interface or at the supplementary material of
our Bioinformatics paper.
What is a KS distribution?¶
KS, also called dS, denotes the synonymous distance between two protein- coding DNA sequences (CDS), where synonymous distance means the expected number of synonymous substitutions per synonymous site. A KS estimate for any two coding sequences can be acquired in two main ways, (1) by means of heuristic counting methods such as the method of Nei & Gojobori (1986) or (2) by using Markov models of codon substitution with Maximum-likelihood (ML) estimation algorithms. The latter methods are most commonly used nowadays, and trace back to the pioneering work of Goldman & Yang (1994) and Muse & Gaut (1994). For more information on the estimation of pairwise synonymous distances, we refer to the wonderful textbooks on molecular evolution authored by professor Ziheng Yang (e.g. Chapter 2 in Yang 2006).
Since synonymous substitutions are presumed to have largely negligible effects on fitness, they are commonly regarded as neutral. Therefore we expect the synonymous distance between two protein coding sequences to increase with time in a stochasti clock like fashion. It is therefore assumed that KS can serve as a proxy for time, and that a larger KS value indicates an older divergence time between two sequences.
A whole paranome KS distribution is nothing more than the distribution of all KS estimates for all inferred duplication events in the genome of some species. It therefore serves as a proxy for the distribution of the divergence times of all duplication events that have left a trace in the genome of interest. Under the assumption of a constant gene duplication rate and a constant rate of loss of duplicated copies, such a distribution is expected to show an exponential decay shape (Lynch & Conery 2000, Lynch 2007), which is the result of a quasi- equilibrium linear birth-death process. A WGD event entails the duplication of all gene copies in the genome at some point in time (i.e. the polyploidization event) and subsequent massive loss or divergence of duplicated copies in a relatively short time frame (i.e. the rediploidization phase). This process of WGD followed by rediploidization is expected to leave a large number of duplication events with similar divergence times (and hence similar KS values) that can be traced back in the extant genome of interest. As a result a WGD is expected to leave a peak signature in the KS distribution that cannot be explained by constant rate small-scale duplication as in the model of Lynch & Conery.
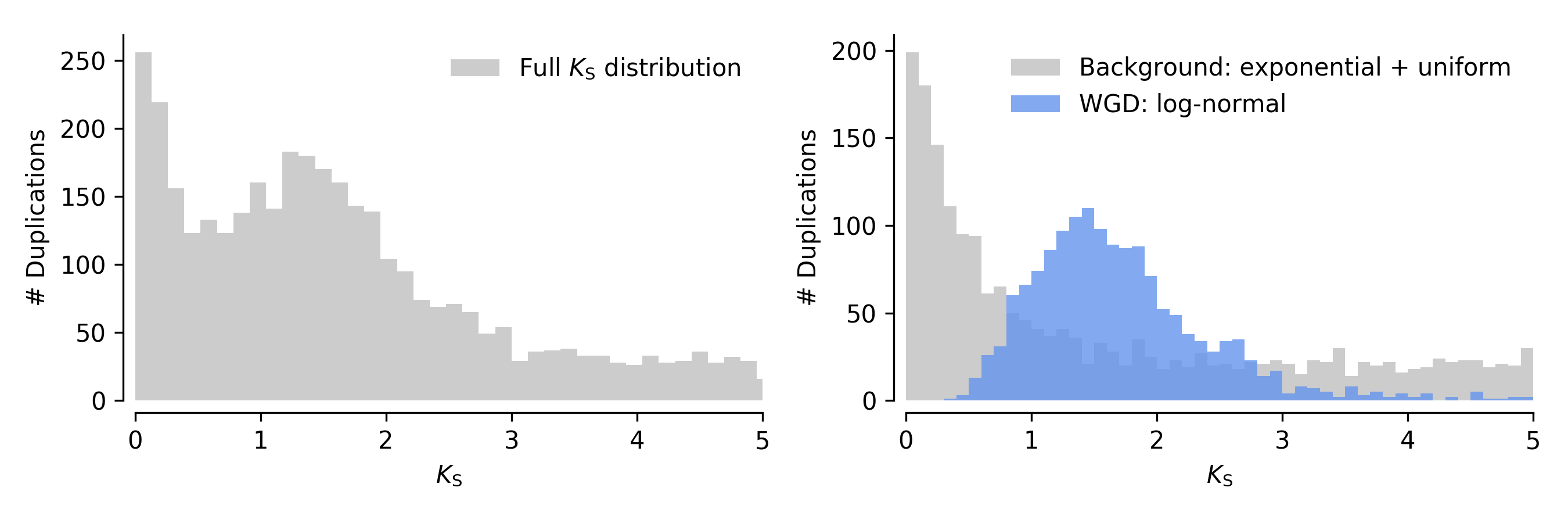
As a side note, we note that if we assume synonymous substitution is a Poisson process, and the synonymous substitution rate is Gamma distributed across gene families, this WGD peak signature is expected to follow a negative binomial distribution in the ditribution of numbers of synonymous substititions (which is not the same as KS distribution). In the KS distribution, this would translate to a peak which has a distribution with positive skew, which we could approximate by a Gamma or Log-Normal distribution. These considerations are important for mixture modeling. An ad hoc illustration of such a hypothetical KS distribution is shown below. The left plot shows the full KS distribution, whereas the right plot shows the decomposition into the background signature from the continuous birth, death and fixation of small-scale duplicates (gray) and the WGD signature (in blue, here modeled as a log-normal distribution).
How is the KS distribution computed in wgd?¶
While the basic idea seems simple enough, constructing a KS
distribution is a rather involved business. This is the main reason why
we released wgd, to make this seemingly simple approach actually
simple to perform. Below we elaborate on the main steps undertken in a
KS distribution construction.
Inferring duplicates¶
First, we need to infer the duplicate genes in the genome. To this end,
commonly employed homolog clustering approaches are used in wgd mcl.
Since we are however not interested in multi-species gene families,
these homolog clustering methods are used only within a particular
genome of interest. wgd mcl clusters genes into paralogous families
(where all genes are paralogous to each other, i.e. diverged after
some duplication event) by
- Doing an all-versus-all Blastp similarity search
- Constructing a sequence similarity graph based on the Blastp results
- Finding clusters in the graph by means of an unsupervised approach using the Markov Clustering algorithm (MCL, Vandongen 2000).
For more information on these approaches, please consult literature on homolog clustering (e.g. OrthoMCL, OrthoFinder), but bear in mind that we are only concerned with within genome homolog clusters, i.e. paralogous gene families.
Calculating pairwise Ks values¶
For every paralogous family we inferred, we first construct a multiple
sequence alignment (MSA) using standard practices (in wgd ksd you
can choose between MUSCLE, MAFFT and PRANK for alignment). Alignments
are constructed at the amino acid level as these can be more reliable,
and are backtranslated to nucleotide sequences to obtain a
codon-alignment.
Now that we have our paralogous gene families and MSAs, we can compute
for every pair in the family the synonymous distance, KS. To this
end wgd ksd uses the codeml program from the PAML package.
codeml has tons of parameter settings and algorithmic options, and
those used in wgd ksd will not be reiterated here but can be found
in the supplement to Zwaenepoel & Van de Peer (2018) or Vanneste et
al. (2013). In general, the settings are set such that they do a good
job at estimating pairwise KS values reliably by maximum likelihood.
Calculating Ks estimates for duplication events¶
Note that after the previous step step we have n(n-1)/2 pairwise KS estimates for a paralogous family with n genes, whereas what we really want is Ks estimate for each duplication event in the family. If multiple duplications happened, we will have multiple redundant KS estimates for internal duplication nodes in the gene family tree. To see this, have a look at the figure below, where a paralogous gene tree is shown, with two marked internal duplication nodes and their respective KS values.
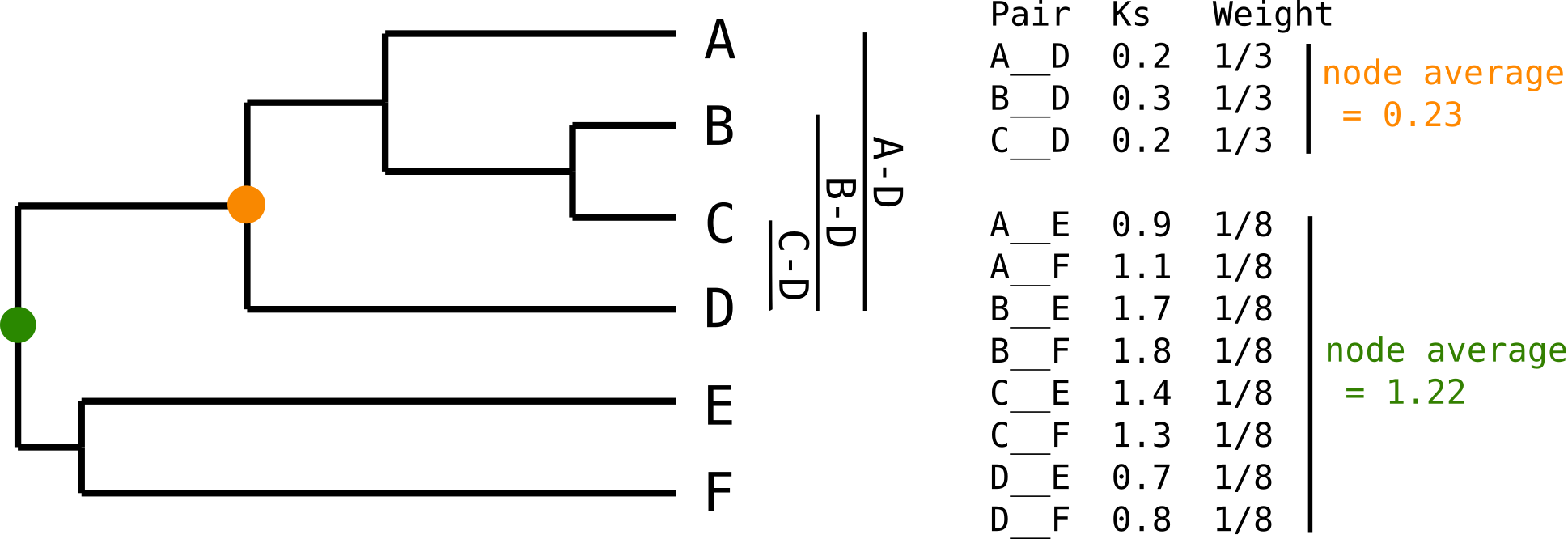
We are interested in the divergence time (approximated by KS) for the five internal nodes of this tree, whereas what we really have is 6x5/2= 15 KS estimates between all pairs of leaves. As is clear from this figure, we have 3 redundant estimates for the orange node, whereas we have 8 redundant estimates for the green node. We can correct for this redundancy by either averaging KS values for each node (or taking some other summary statistic like the median), or by computing a weight for every KS value. (see also next section).
However, to do this we of course need the tree, therefore wgd ksd will
compute for every paralogous gene family a phylogenetic tree after calculating
the pairwise KS values to do redundancy correction (either by node-averaging
or node-weighting, see below). In wgd ksd one can choose between three
options to construct this tree, either average linkage clustering of KS
values (which is fast but somewhat crude), approximate ML tree inference using
FastTree (Price et al.) or ML tree inference using PhyML (which is obviously
the slowest option).
After the tree is computed wgd ksd will write for every pair the Ks
estimate, some alignment statistics, the node in the tree for which the Ks
value provides a divergence estimate, the gene family, the various distance
estimates (KS, KA and ω) and the weights computed without filtering out
outliers and with filtering out outliers (see below).
Visualizing a KS distribution¶
If all goes well, wgd ksd will compute a KS distribution which looks
somewhat like this (where I have omitted lots of columns that are not really
of interest currently):
Family Ks Node WeightOutliersExcluded
AT3G11180__AT5G05600 GF_000093 0.7679 23.0 1.00
AT2G38240__AT5G05600 GF_000093 1.9122 25.0 0.33
AT2G38240__AT3G11180 GF_000093 2.5219 25.0 0.33
AT5G08640__AT5G63580 GF_000093 1.3265 30.0 0.20
... ...
Now, simply plotting a histogram of the resulting KS values will of course be flawed, since we will plot all redundant estimates with equal weight, and since older duplication events will have more pairwise estimates, this may artificially amplify the signal of old duplication events.
There are two main approaches to overcome this, already noted in the previous
section. The first is node-averaging, where one computes a summarized KS
value for each node. This is easily performed using built-in functions to work
with data frames in R or Python. This approach obviously throws away the
information in the individual pairwise estimates and only uses the summarized
value in subsequent analysis. The second approach is node-weighting, where
one computes a weight for every pairwise estimate, and adds every single
estimate to the histogram but with some weighted based on the number of
redundant etimates. Such an approach does not throw away the information in
individual estimates, but is trickier to work with in subsequent analyses such
as Kernel density estimation (KDE) or Gaussian mixture modeling (GMMs).
Weighted histograms can be plotted both in ggplot2 in R and matplotlib
in Python.
It is important to be aware that different filters on the KS range, will
cause the weights to change. Indeed, if some pairwise estimate is filtered out,
because of some filtering riterion, the number of KS estimates will change
for that particular duplication event. This is exactly the difference between
the WeightOutliersIncluded column and WeightOutliersExcluded column, where
the weights in the latter column are based on the number of pairwise estimates
after filtering, whereas the former has weights based on the number of pairwise
estimates before filtering. It is does important to recompute the weights when
different filtering riteria are used. The tools in wgd will always do this,
but it is important to keep in mind when analyzing the dat yourelf in R or some
other statistical/plotting environment.
Note that wgd ksd by default outputs node-weighted histograms, whereas
node-averaged histograms can be generated using wgd viz. The mixture
modeling and KDE methods in wgd mix and wgd kde use node-averaged
histograms for modeling purposes.
Downstream analyses¶
Downstream analyses consist mainly of fitting different models to a KS distribution, which could be KDEs, or mixture models. For more information on mixture modeling of KS distributions, we refer to A note on mixture models for KS distributions.