wgd: Whole genome duplication analysis in Python¶
This Python package and corresponding command line interface (CLI) were
developed for various analyses related to whole genome duplications (WGDs).
Here the Python API is documented as well as the various command line
utilities bundled in the wgd CLI.
Installation¶
To install wgd, go to the repository available at
https://github.com/arzwa/wgd and follow installation instructions there.
Example & information¶
- To get started with
wgdhead straight to the command line interface page. - For more information on KS distributions and how
wgdcomputes them have a look at these notes. - For an additional step by step example, we refer to the supplementary information of our Bioinformatics Applications note paper (open access) at https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/bty915/5162749
External software¶
wgd requires the following third party executables (preferably these should
also be in the PATH environment variable):
For wgd blast:
- BLAST, from which it uses the
blastpandmakeblastdbcommands,sudo apt-get install ncbi-blast+will often suffice for installation - MCL (https://micans.org/mcl/index.html). Get MCL using your package
manager
sudo apt-get install mclor download it at the provided link.
For wgd ks (most of these can also be installed with apt-get):
- One of the following multiple sequence alignment programs: MUSCLE, MAFFT or
PRANK (
sudo apt-get install muscle mafft prank) - CODEML from the PAML software package (Yang 1997). PAML can be downloaded
from the following link: http://abacus.gene.ucl.ac.uk/software/paml.html. It
can also be installed using
sudo apt-get install paml. - For node weighting using phylogenetic trees, PhyML and FastTree are supported,
but average linkage clustering (no external software needed) can be used as an
alternative. To install
sudo apt-get install fasttree phyml. (Note that FastTree should be executable asFastTreeand notfasttree, so please specify an alias or symlink from the latter to the former if necessary.)
For wgd syn
- i-ADHoRe 3.0 suite (http://bioinformatics.psb.ugent.be/beg/tools/i-adhore30)
Of course, you don’t need all of these tools installed for each feature of
wgd.
Command line tools¶
The command line tools are the main functionality of the wgd package.
You can find tools for the following analyses:
- All-versus-all Blastp analysis and MCL clustering
- Whole paranome KS (and KA and ω) distribution construction
- One-versus-one orthologs KS (and KA and ω;) distribution construction
- Mixture modeling of KS distributions and WGD-specific paralog extraction
- Interactive visualization of (multiple) KS distributions and kernel density estimates thereof
- Intragenomic co-linearity/synteny analysis and anchor based KS distribution construction
- Co-linearity dotplot construction
All information can be found here:
Here is a flow chart of the different analyses that can be performed using the wgd CLI:
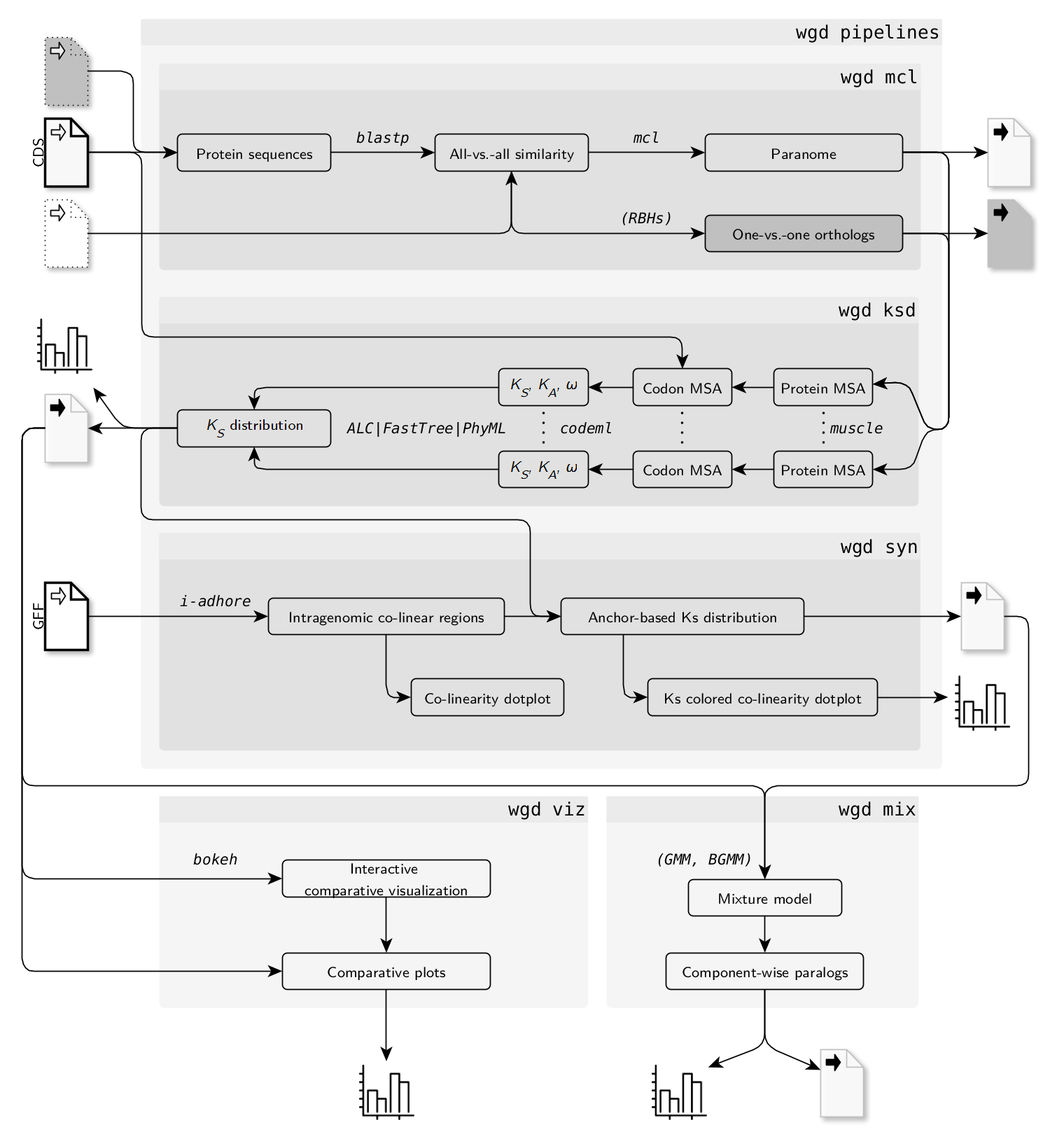
Citation¶
If you use wgd, please cite:
- Zwaenepoel, A., and Van de Peer, Y. wgd - simple command line tools for
the analysis of ancient whole genome duplications. Bioinformatics., bty915,
https://doi.org/10.1093/bioinformatics/bty915
Furthermore, for the specific tools in wgd, please cite the following:
If you use wgd mcl please cite:
- Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W.,
and Lipman, D. J. (1997). Gapped BLAST and PSI-BLAST: a new generation of
protein database search programs. Nucleic Acids Research, 25(17), 3389–3402.
- van Dongen, S. (2000). Graph Clustering by Flow Simulation. Ph.D. thesis,
University of Utrecht, Utrecht.
For wgd ksd, please cite:
- Yang, Z. (2007). PAML 4: Phylogenetic Analysis by Maximum Likelihood.
Molecular Biology and Evolution, 24(8), 1586–1591.
- [if using MUSCLE] Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and
high throughput. Nucleic Acids Research, 32(5), 1792–1797.
- [if using MAFFT] Katoh, K. and Standley, D. M. (2013). MAFFT multiple sequence alignment software
version 7: improvements in performance and usability. Molecular Biology and
Evolution, 30(4), 772–780.
- [if using PRANK] Löytynoja, A. and Goldman, N. (2008). Phylogeny-Aware Gap Placement Prevents
Errors in Sequence Alignment and Evolutionary Analysis. Science, 320(5883),
1632–1635.
- [if using FastTree] 2825–2830.
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2 - Approximately
Maximum-Likelihood Trees for Large Alignments. PLOS ONE, 5(3), e9490.
- [if using PhyML] Guindon, S., Dufayard, J.-F., Lefort, V., Anisimova, M., Hordijk, W., and
Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood
phylogenies: assessing the performance of PhyML 3.0. Systematic Biology, 59(3),
307–321.
For wgd syn, please cite:
- Proost, S., Fostier, J., De Witte, D., Dhoedt, B., Demeester, P., Van de Peer, Y., and
Vandepoele, K. (2012). i-ADHoRe 3.0 : fast and sensitive detection of genomic
homology in extremely large data sets. NUCLEIC ACIDS RESEARCH, 40(2).
Python package¶
For those interested in the underlying structure of wgd, here you can find
the full documentation of the API.
Contents: